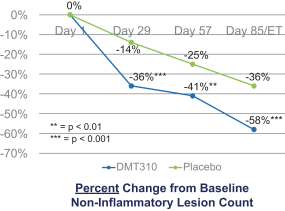
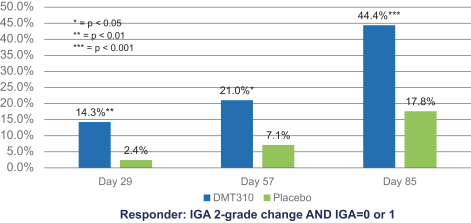
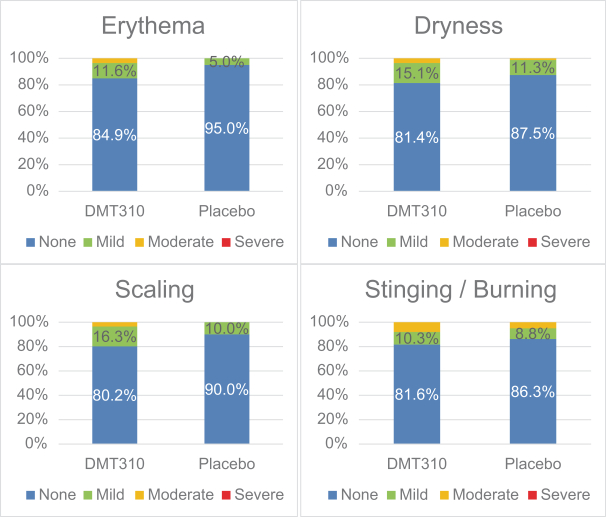
The information contained in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
| PRELIMINARY PROSPECTUS | SUBJECT TO COMPLETION | DATED , 2021 |
Shares
Common Stock

Dermata Therapeutics, Inc.
This is a firm commitment initial public offering of shares of Dermata Therapeutics, Inc. common stock. Prior to this offering, there has been no public market for our common stock. We anticipate that the initial public offering price of our common stock will be between $ and $ per share.
We have applied to list our shares of common stock for trading on the Nasdaq Capital Market under the symbol “DRMA.” No assurance can be given that our application will be approved. If our common stock is not approved for listing on the NASDAQ Capital Market, we will not consummate this offering.
We are an “emerging growth company” under the Jumpstart our Business Startups Act of 2012, or JOBS Act, and, as such, have elected to comply with certain reduced public company reporting requirements for this prospectus and future filings.
Investing in our common stock is highly speculative and involves a high degree of risk. See “Risk Factors” beginning on page 10 of this prospectus for a discussion of information that should be considered in connection with an investment in our common stock.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
| Per Share | Total | |||||||
| Initial public offering price |
$ | $ | ||||||
| Underwriting discounts and commissions (1) |
$ | $ | ||||||
| Proceeds to us, before expenses |
$ | $ | ||||||
| (1) | Does not include the reimbursement of certain expenses of the underwriters. We refer you to “Underwriting” beginning on page 144 for additional information regarding the underwriters’ compensation. |
The underwrites may also exercise their option to purchase up to additional shares of common stock at the public offering price per share, less the underwriting discount, for 45 days after the date of this prospectus to cover over-allotments, if any. If the underwriters exercise this option in full, the total underwriting discounts and commissions will be $ and the additional proceeds to us, before expenses, from the over-allotment option exercise will be $ .
The underwriters expect to deliver our shares of common stock in the offering on or about , 2021.
Book Running Manager
Maxim Group LLC
The date of this prospectus is , 2021