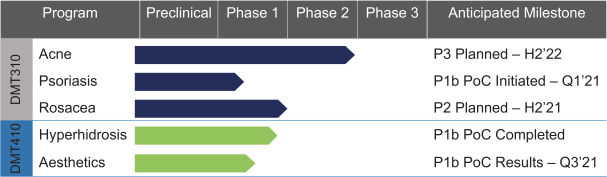
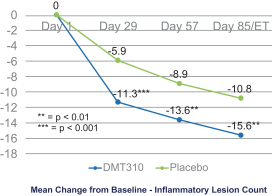
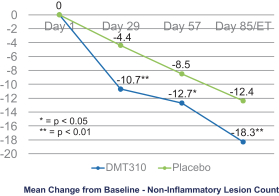
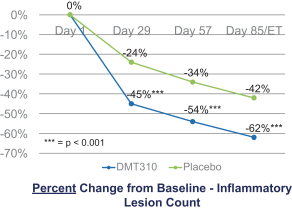
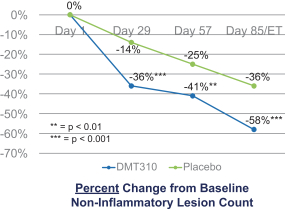
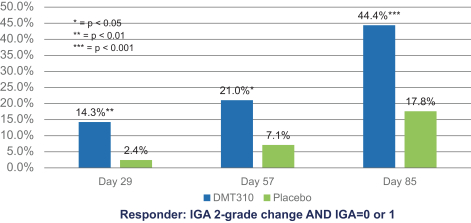
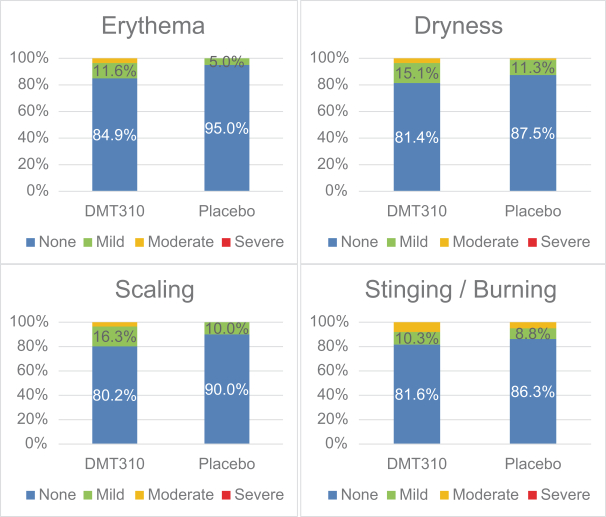
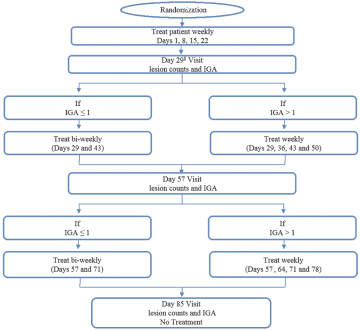
The information contained in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
| PRELIMINARY PROSPECTUS | SUBJECT TO COMPLETION | DATED August 6, 2021 |
1,875,000 Units consisting of
1,875,000 Shares of Common Stock and
Warrants to purchase up to 1,875,000 Shares of Common Stock

Dermata Therapeutics, Inc.
Dermata Therapeutics, Inc. is offering units, or Units, each consisting of one share of our common stock and one warrant to purchase one share of our common stock, or each, a Warrant, in an initial public offering. No public market currently exists for our common stock or the Warrants comprising the Units. The estimated initial public offering price per Unit is between $7.00 and $9.00 per share, and the number of Units offered hereby is based upon an assumed offering price of $8.00 per Unit, the midpoint of such estimated price range. The Units have no stand-alone rights and will not be certificated or issued as stand-alone securities. The common stock and Warrants are immediately separable and will be issued separately in this offering. Each Warrant offered hereby is immediately exercisable on the date of issuance at an exercise price of $ per share of common stock (which shall not be less than 100% of the public offering price per Unit), and will expire five years from the date of issuance.
We have applied to list our common stock and Warrants on the Nasdaq Capital Market, or Nasdaq, under the symbols “DRMA” and “DRMAW,” respectively. One of the requirements for listing on the Nasdaq Capital Market is the minimum market value of publicly traded shares of $15,000,000. In order to meet such requirement, we expect to sell no less than $15,000,000 of unrestricted securities in this offering. No assurance can be given that our application will be approved. If our common stock and Warrants are not approved for listing on Nasdaq, we will not consummate this offering.
We are an “emerging growth company” under the Jumpstart our Business Startups Act of 2012, or JOBS Act, and, as such, have elected to comply with certain reduced public company reporting requirements for this prospectus and future filings.
Investing in our securities is highly speculative and involves a high degree of risk. See “Risk Factors” beginning on page 12 of this prospectus for a discussion of information that should be considered in connection with an investment in our securities.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
| Per Unit |
Total | |||||||
| Initial public offering price |
$ | $ | ||||||
| Underwriting discounts and commissions (1) |
$ | $ | ||||||
| Proceeds to us, before expenses |
$ | $ | ||||||
| (1) | Does not include the reimbursement of certain expenses of the underwriters. We refer you to “Underwriting” beginning on page 152 for additional information regarding the underwriters’ compensation. |
We have granted the representative of the underwriters an option, exercisable within 45 days from the date of this prospectus, to purchase from us, up to an additional 281,250 shares of common stock at a per share price of $ and/or up to an additional 281,250 Warrants to purchase up to 281,250 shares of common stock at a price per Warrant of $ , less, in each case, the underwriting discounts and commissions, to cover over-allotments, if any. If the representative of the underwriters exercises the option in full, the total underwriting discounts and commissions payable will be $ , and the total proceeds to us, before expenses, will be $ .
Certain of our existing stockholders and their affiliated entities that are affiliated with our founder and Chief Executive Officer and certain of our current directors, including Proehl Investment Ventures, LLC, and Hale BioPharma Ventures, LLC, have indicated an interest in purchasing up to an aggregate of approximately $1.25 million worth of Units in this offering at the initial public offering price. However, because indications of interest are not binding agreements or commitments to purchase, the underwriters could determine to sell more, less or no Units to any of these stockholders and any of these stockholders could determine to purchase more, less or no Units in this offering. The underwriters will receive the same underwriting discounts and commissions on any Units purchased by these stockholders as they will on any other Units sold to the public in this offering.
The underwriters expect to deliver the securities comprising the Units on or about , 2021.
| Sole Book Running Manager Maxim Group LLC |
| Co-Manager Brookline Capital Markets, a division of Arcadia Securities, LLC |
The date of this prospectus is , 2021